ACSF3
Acyl-CoA-Synthetase-Familienmitglied 3 (ACSF3) ist ein mitochondriales Enzym, das beim Menschen durch das Gen ACSF3 kodiert wird.[1] Das Enzym gehört zur Familie der Acyl-CoA-Synthetasen.[2]
Struktur
Das Gen ACSF3 befindet sich auf dem 16. Chromosom, und zwar auf der Position 16q24.3. Das Gen enthält 17 Exons.[1] ACSF3 kodiert ein 64,1 kDa großes Protein, das sich aus 576 Aminosäuren zusammensetzt, 20 Peptide wurden durch Massenspektrometrie-Daten beobachtet.[3][4]
Mögliche posttranslationale Modifikationen an ACSF3:
- Acetylierung an Lysin 565 (K565)[5]
Funktion
Dieses Gen kodiert ein Mitglied der Acyl-CoA-Synthetase-Familie von Enzymen, die Fettsäuren aktivieren, indem sie die Bildung einer Thioesterbindung zwischen Fettsäuren und Coenzym A katalysieren. Das kodierte Protein ist in den Mitochondrien lokalisiert, hat eine hohe Spezifität für Malonsäure und Methylmalonsäure und besitzt Malonyl-CoA-Synthetase-Aktivität:[1][6]
- ATP + Malonat + CoA AMP + Diphosphat + Malonyl-CoA

ACSF3 wandelt am effizientesten Malonat in Malonyl-CoA um, aber auch Methylmalonat wird mit etwa 70 % dieser Aktivität effektiv aktiviert.[7]
Abbau von Malonsäure und Methylmalonsäure
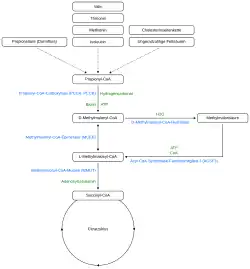
Durch die Umwandlung von Malonat in Malonyl-CoA spielt ACSF3 eine entscheidende Rolle bei der Entfernung von intramitochondrialem Malonat, einem starken Inhibitor der mitochondrialen Atmung.[8][9] Malonat hemmt kompetitiv die Succinat-Dehydrogenase (SDH), ein Enzym, das sowohl im Citratzyklus als auch als Komplex II der Atmungskette wirkt.[8][9] Dagegen beeinträchtigt Methylmalonat die Aktivität der SDH indirekt, indem es den mitochondrialen Succinat-Import stört, anstatt das Enzym direkt zu hemmen.[10] Die Beseitigung beider Säuren ist nicht nur entscheidend, um eine mitochondriale Dysfunktion zu verhindern, sondern auch, um eine metabolische Azidose zu vermeiden.[11][12] Laut einem Acsf3-Knockout-Mausmodell wurde der Threonin-Katabolismus als Hauptursache für die Anhäufung von Methylmalonsäure identifiziert.[11]
Synthese von mitochondrialem Malonyl-CoA
Mitochondriale Fettsäuresynthese (mtFAS)
_pathway.svg.png)
Malonyl-CoA, das einerseits durch ACSF3 und andererseits durch die mitochondriale Isoform der Acetyl-CoA-Carboxylase 1 (mtACC1) aus Acetyl-CoA gebildet wird, liefert die Verlängerungseinheiten für die mitochondriale Fettsäuresynthese (mtFAS).[13] Die daraus entstehenden Acyl-ACP-Varianten erfüllen je nach Kettenlänge unterschiedliche Funktionen: So wird beispielsweise Octanoyl-ACP (C8) für die Biosynthese von Liponsäure benötigt, einem Cofaktor wichtiger mitochondrialer Enzymkomplexe wie dem Pyruvat-Dehydrogenase-Komplex (PDC), dem α-Ketoglutarat-Dehydrogenase-Komplex (OGDC), dem 2-Oxoadipat-Dehydrogenase-Komplex (OADHC), dem verzweigtkettigen α-Ketosäure-Dehydrogenase-Komplex (BCKDHC) und dem Glycine-Cleavage-System (GCS).[14] Längerkettige Varianten (C10–16) hingegen aktivieren allosterisch das Netzwerk der LYRM-Proteine.[15][16] Beim Menschen umfasst dieses Netzwerk mindestens 12 Proteine und reguliert die mitochondriale Translation, die Biogenese von Eisen-Schwefel-Clustern sowie die Assemblierung der Komplexe der Atmungskette.[17][16]
Proteinmalonylierung
Neben seiner Rolle in der mtFAS ist Malonyl-CoA auch essentiell für die Lysin-Malonylierung mitochondrialer Proteine, einer posttranslationale Modifikation, die zur metabolischen Effizienz in Säugetierzellen beiträgt.[8] ACSF3 reguliert die rhythmische Malonylierung mitochondrialer Proteine in Abhängigkeit von der Nahrungsaufnahme und beeinflusst dabei hepatische Stoffwechselwege wie die Glykogenmobilisierung, Lipidsynthese und Triglyceridansammlung.[18] Allerdings scheinen die Lysin-Malonylierungsspiegel bei einer Hemmung von ACSF3 zelltypspezifisch beeinflusst zu werden.[11]
Synthese von Acetyl-CoA
Außerdem kann Malonyl-CoA durch die Malonyl-CoA-Decarboxylase (MLYCD) in Acetyl-CoA umgewandelt werden, das anschließend in den Citratzyklus eingespeist wird.[18]
Klinische Bedeutung
Kombinierte Malon- und Methylmalonazidurie (CMAMMA)
Es hat sich gezeigt, dass Mutationen im ACSF3-Gen die Stoffwechselerkrankung kombinierte Malon- und Methylmalonazidurie (CMAMMA) verursachen.[12] CMAMMA ist eine Erkrankung, die durch hohe Konzentrationen von Methylmalonsäure und Malonsäure gekennzeichnet ist, da ein Mangel in diesem Gen dazu führt, dass diese Metaboliten nicht abgebaut werden können. Die Krankheit wird in der Regel entweder durch einen Gentest oder durch höhere Methylmalonsäure- als Malonsäurespiegel diagnostiziert, auch wenn beide Werte erhöht sind. Durch die Berechnung des Verhältnisses von Malonsäure zu Methylmalonsäure im Blutplasma kann CMAMMA von der klassischen Methylmalonazidurie unterschieden werden.[19] Die Störung zeigt typischerweise Symptome in der frühen Kindheit, die zunächst mit hohen Säurewerten im Blut (Ketoazidose) beginnen. Die Erkrankung kann sich auch in Form von unwillkürlichen Muskelverspannungen (Dystonie), schwachem Muskeltonus (Muskelhypotonie), Entwicklungsverzögerung, Unfähigkeit in der erwarteten Geschwindigkeit zu wachsen und an Gewicht zuzunehmen (Gedeihstörung), Unterzuckerung (Hypoglykämie) und Koma äußern. Bei einigen betroffenen Kindern kann sogar eine Mikrozephalie auftreten. Andere Menschen mit CMAMMA entwickeln bis zum Erwachsenenalter keine Anzeichen und Symptome. Diese Menschen haben in der Regel neurologische Probleme wie Krampfanfälle, Gedächtnisverlust, eine Abnahme der geistigen Leistungsfähigkeit oder psychiatrische Erkrankungen.[1]
Chronisch obstruktive Lungenerkrankung (COPD)
Eine epigenetische Studie fand eine unterschiedliche DNA-Methylierung des ACSF3-Gens im fetalen Lungengewebe, das mütterlichem Tabakkonsum ausgesetzt war, was auf eine mögliche Rolle bei der Entstehung der chronisch obstruktiven Lungenerkrankung (COPD) bereits während der Entwicklung hindeutet.[20] Darüber hinaus haben integrative Analysen der DNA-Methylierung und Genexpression in Lungengewebe ACSF3 als einen Schlüsselfaktor bei der Regulation von COPD identifiziert.[20]
Metabolische Dysfunktion-assoziierte steatotische Lebererkrankung (MASLD)
ACSF3 ist an der Pathophysiologie der mit metabolischer Dysfunktion assoziierten steatotischen Lebererkrankung (MASLD, früher NAFLD) beteiligt.[5] Seine Expression ist bei Mausmodellen mit fettreicher Ernährung sowie bei den Erkrankungen Adipositas und alkoholischer Lebererkrankung erhöht, wobei letztere beiden mit einer Beeinträchtigung des mitochondrialen Fettsäurestoffwechsels und einer verstärkten Lipidperoxidation einhergehen.[5] Die Deacetylierung von ACSF3 durch die mitochondriale Deacetylase Sirtuin 3 (SIRT3) führt zu einer verringerten Stabilität und fördert den Abbau von ACSF3, was unter Bedingungen einer fettreichen Ernährung die hepatische Lipidhomöostase verbessert und die Steatose in Mausmodellen reduziert.[5] Es konnte gezeigt werden, dass die phenolische Verbindung Protocatechusäure (PCA) SIRT3 aktiviert, was die SIRT3-ACSF3-Achse als möglichen therapeutischen Ansatzpunkt für MASLD in den Blickpunkt rückt.[5][21]
Evolutionäre Rolle
Eine uralte, menschenspezifische regulatorische Variante, rs34590044-A, erhöht die Expression des ACSF3-Gens und fördert dadurch eine größere Körpergröße sowie einen höheren Grundumsatz (Basal Metabolic Rate, BMR).[11] Im Vergleich zu nicht-menschlichen Menschenaffen zeigen anatomisch moderne Menschen sowohl eine höhere ACSF3-Expression als auch einen erhöhten Grundumsatz, was auf eine evolutionäre Veränderung hinweist.[11] Diese Variante hat vermutlich die Anpassung an fleischreiche Ernährung begünstigt, insbesondere durch einen verbesserten Threoninstoffwechsel.[11] Studien an Mäusen und Zellkulturen zeigen, dass ACSF3 die mitochondriale Funktion und das Knochenwachstum unterstützt, während eine Störung von ACSF3 beides beeinträchtigt.[11] Die Ergebnisse bringen ACSF3 mit der Koevolution von Körpergröße, Stoffwechsel und Ernährungsanpassungen in der menschlichen Evolution in Verbindung.[11]
Siehe auch
Einzelnachweise
- ↑ a b c d Entrez Gene: Acyl-CoA synthetase family member 3. Abgerufen am 30. Dezember 2011.
- ↑ Paul A. Watkins, Dony Maiguel, Zhenzhen Jia, Jonathan Pevsner: Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. In: Journal of Lipid Research. Band 48, Nr. 12, Dezember 2007, S. 2736–2750, doi:10.1194/jlr.M700378-JLR200 (elsevier.com).
- ↑ Nobel C. Zong, Haomin Li, Hua Li, Maggie P.Y. Lam, Rafael C. Jimenez, Christina S. Kim, Ning Deng, Allen K. Kim, Jeong Ho Choi, Ivette Zelaya, David Liem, David Meyer, Jacob Odeberg, Caiyun Fang, Hao-jie Lu, Tao Xu, James Weiss, Huilong Duan, Mathias Uhlen, John R. Yates, Rolf Apweiler, Junbo Ge, Henning Hermjakob, Peipei Ping: Integration of Cardiac Proteome Biology and Medicine by a Specialized Knowledgebase. In: Circulation Research. Band 113, Nr. 9, 12. Oktober 2013, ISSN 0009-7330, S. 1043–1053, doi:10.1161/CIRCRESAHA.113.301151, PMID 23965338, PMC 4076475 (freier Volltext) – (ahajournals.org).
- ↑ Acyl-CoA synthetase family member 3, mitochondrial. In: Cardiac Organellar Protein Atlas Knowledgebase (COPaKB). Ehemals im (nicht mehr online verfügbar); abgerufen am 25. März 2015. (Seite nicht mehr abrufbar. Suche in Webarchiven)
- ↑ a b c d e Ruimin Sun, Xiaohui Kang, Yan Zhao, Zhanyu Wang, Ruiwen Wang, Rong Fu, Yang Li, Yan Hu, Zhecheng Wang, Wen Shan, Junjun Zhou, Xiaofeng Tian, Jihong Yao: Sirtuin 3‐mediated deacetylation of acyl‐ CoA synthetase family member 3 by protocatechuic acid attenuates non‐alcoholic fatty liver disease. In: British Journal of Pharmacology. Band 177, Nr. 18, September 2020, ISSN 0007-1188, S. 4166–4180, doi:10.1111/bph.15159, PMID 32520409, PMC 7443473 (freier Volltext) – (wiley.com [abgerufen am 28. Juni 2025]).
- ↑ ExplorEnz: EC 6.2.1.76. Abgerufen am 27. April 2025.
- ↑ Andrzej Witkowski, Jennifer Thweatt, Stuart Smith: Mammalian ACSF3 Protein Is a Malonyl-CoA Synthetase That Supplies the Chain Extender Units for Mitochondrial Fatty Acid Synthesis. In: Journal of Biological Chemistry. Band 286, Nr. 39, September 2011, S. 33729–33736, doi:10.1074/jbc.M111.291591, PMID 21846720, PMC 3190830 (freier Volltext) – (elsevier.com).
- ↑ a b c Caitlyn E. Bowman, Susana Rodriguez, Ebru S. Selen Alpergin, Michelle G. Acoba, Liang Zhao, Thomas Hartung, Steven M. Claypool, Paul A. Watkins, Michael J. Wolfgang: The Mammalian Malonyl-CoA Synthetase ACSF3 Is Required for Mitochondrial Protein Malonylation and Metabolic Efficiency. In: Cell Chemical Biology. Band 24, Nr. 6, Juni 2017, S. 673–684.e4, doi:10.1016/j.chembiol.2017.04.009, PMID 28479296, PMC 5482780 (freier Volltext).
- ↑ a b Caitlyn E. Bowman, Michael J. Wolfgang: Role of the malonyl-CoA synthetase ACSF3 in mitochondrial metabolism. In: Advances in Biological Regulation. Band 71, Januar 2019, S. 34–40, doi:10.1016/j.jbior.2018.09.002, PMID 30201289, PMC 6347522 (freier Volltext) – (elsevier.com).
- ↑ S. R. Mirandola, D. R. Melo, P. F. Schuck, G. C. Ferreira, M. Wajner, R. F. Castilho: Methylmalonate inhibits succinate‐supported oxygen consumption by interfering with mitochondrial succinate uptake. In: Journal of Inherited Metabolic Disease. Band 31, Nr. 1, Februar 2008, ISSN 0141-8955, S. 44–54, doi:10.1007/s10545-007-0798-1 (wiley.com).
- ↑ a b c d e f g h Yufeng Zhang, Jie Wang, Chuanyou Yi, Yue Su, Zi Yin, Shuxian Zhang, Li Jin, Mark Stoneking, Jian Yang, Ke Wang, He Huang, Jin Li, Shaohua Fan: An ancient regulatory variant of ACSF3 influences the coevolution of increased human height and basal metabolic rate via metabolic homeostasis. In: Cell Genomics. Band 5, Nr. 6, Juni 2025, S. 100855, doi:10.1016/j.xgen.2025.100855 (elsevier.com).
- ↑ a b A. Alfares, L. D. Nunez, K. Al-Thihli, J. Mitchell, S. Melancon, N. Anastasio, K. C. H. Ha, J. Majewski, D. S. Rosenblatt, N. Braverman: Combined malonic and methylmalonic aciduria: exome sequencing reveals mutations in the ACSF3 gene in patients with a non-classic phenotype. In: Journal of Medical Genetics. Band 48, Nr. 9, 1. September 2011, ISSN 0022-2593, S. 602–605, doi:10.1136/jmedgenet-2011-100230 (bmj.com).
- ↑ Geoffray Monteuuis, Fumi Suomi, Juha M. Kerätär, Ali J. Masud, Alexander J. Kastaniotis: A conserved mammalian mitochondrial isoform of acetyl-CoA carboxylase ACC1 provides the malonyl-CoA essential for mitochondrial biogenesis in tandem with ACSF3. In: Biochemical Journal. Band 474, Nr. 22, 15. November 2017, ISSN 0264-6021, S. 3783–3797, doi:10.1042/BCJ20170416 (portlandpress.com).
- ↑ Zeinab Wehbe, Sidney Behringer, Khaled Alatibi, David Watkins, David Rosenblatt, Ute Spiekerkoetter, Sara Tucci: The emerging role of the mitochondrial fatty-acid synthase (mtFASII) in the regulation of energy metabolism. In: Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. Band 1864, Nr. 11, November 2019, ISSN 1388-1981, S. 1629–1643, doi:10.1016/j.bbalip.2019.07.012.
- ↑ Jia Xin Tang, Kyle Thompson, Robert W. Taylor, Monika Oláhová: Mitochondrial OXPHOS Biogenesis: Co-Regulation of Protein Synthesis, Import, and Assembly Pathways. In: International Journal of Molecular Sciences. Band 21, Nr. 11, 28. Mai 2020, ISSN 1422-0067, S. 3820, doi:10.3390/ijms21113820, PMID 32481479, PMC 7312649 (freier Volltext) – (mdpi.com [abgerufen am 28. Juni 2025]).
- ↑ a b Jonathan G. Van Vranken, Sara M. Nowinski, Katie J. Clowers, Mi-Young Jeong, Yeyun Ouyang, Jordan A. Berg, Jeremy P. Gygi, Steven P. Gygi, Dennis R. Winge, Jared Rutter: ACP Acylation Is an Acetyl-CoA-Dependent Modification Required for Electron Transport Chain Assembly. In: Molecular Cell. Band 71, Nr. 4, August 2018, S. 567–580.e4, doi:10.1016/j.molcel.2018.06.039, PMID 30118679, PMC 6104058 (freier Volltext) – (elsevier.com).
- ↑ Ali J. Masud, Alexander J. Kastaniotis, M. Tanvir Rahman, Kaija J. Autio, J. Kalervo Hiltunen: Mitochondrial acyl carrier protein (ACP) at the interface of metabolic state sensing and mitochondrial function. In: Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. Band 1866, Nr. 12, Dezember 2019, S. 118540, doi:10.1016/j.bbamcr.2019.118540 (elsevier.com).
- ↑ a b Enora Le Questel, Charlène Besnard, Florian Atger, Yolène Foucher, Alwéna Tollec, Victoria Pakulska, Arsênio Rodrigues Oliveira, Chloé Clotteau, Mathilde Gourdel, Ivan Nemazanyy, Mikael Croyal, Yohann Coute, David Jacobi, Bertrand Cariou, Daniel Mauvoisin: Diurnal regulation of Acyl-CoA synthetase 3 (ACSF3) underlies daily mitochondrial lysine-malonylation and hepatic metabolism. 6. September 2024, abgerufen am 27. April 2025 (englisch).
- ↑ Monique G. M. de Sain-van der Velden, Maria van der Ham, Judith J. Jans, Gepke Visser, Hubertus C. M. T. Prinsen, Nanda M. Verhoeven-Duif, Koen L. I. van Gassen, Peter M. van Hasselt: A New Approach for Fast Metabolic Diagnostics in CMAMMA. In: JIMD Reports, Volume 30. Band 30. Springer Berlin Heidelberg, Berlin, Heidelberg 2016, ISBN 978-3-662-53680-3, S. 15–22, doi:10.1007/8904_2016_531, PMID 26915364, PMC 5110436 (freier Volltext) – (springer.com [abgerufen am 24. Februar 2024]).
- ↑ a b Jonas Eriksson Ström, Simon Kebede Merid, Robert Linder, Jamshid Pourazar, Anne Lindberg, Erik Melén, Annelie F. Behndig: Airway MMP-12 and DNA methylation in COPD: an integrative approach. In: Respiratory Research. Band 26, Nr. 1, 10. Januar 2025, ISSN 1465-993X, doi:10.1186/s12931-024-03088-3, PMID 39794761, PMC 11724436 (freier Volltext) – (biomedcentral.com).
- ↑ Shengyu Zhang, Congcong Shen, Han Di, Yanhong Wang, Feng Guan: Regulatory Mechanisms of Phenolic Acids in Metabolic Dysfunction-Associated Steatotic Liver Disease: A Review. In: Antioxidants. Band 14, Nr. 7, 20. Juni 2025, ISSN 2076-3921, S. 760, doi:10.3390/antiox14070760 (mdpi.com).
Literaturhinweise
- P. A. Watkins, D. Maiguel, Z. Jia, J. Pevsner: Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. In: Journal of Lipid Research. Band 48, Nr. 12, Dezember 2007, S. 2736–2750. doi:10.1194/jlr.M700378-JLR200. PMID 17762044.
Weblinks
Dieser Artikel enthält einen Text aus der United States National Library of Medicine, der gemeinfrei ist.