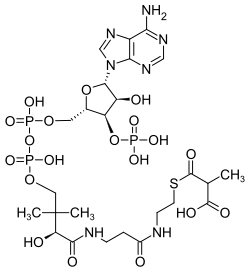
Methylmalonyl-CoA
| Strukturformel | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||
| Allgemeines | |||||||||||||
| Name | Methylmalonyl-CoA | ||||||||||||
| Summenformel | C25H40N7O19P3S | ||||||||||||
| Externe Identifikatoren/Datenbanken | |||||||||||||
| |||||||||||||
| Eigenschaften | |||||||||||||
| Molare Masse | 867,6 g·mol−1 | ||||||||||||
| Sicherheitshinweise | |||||||||||||
| |||||||||||||
| Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen (0 °C, 1000 hPa). | |||||||||||||
Methylmalonyl-Coenzym A, kurz Methylmalonyl-CoA, ist eine organische chemische Verbindung. Sie ist ein Thioester – aus Coenzym A und Methylmalonsäure – sowie eine Carbonsäure. In der Biochemie wird auch das Anion der Carbonsäure als Methylmalonyl-CoA bezeichnet. Der Thioester tritt als Zwischenprodukt beim Abbau von Fettsäuren mit ungerader Anzahl von Kohlenstoff-Atomen[2] sowie einiger Aminosäuren[3] auf. Das beim Abbau von Methylmalonyl-CoA gebildete Produkt Succinyl-CoA entsteht im Metabolismus auch im Citratzyklus; die Reaktion mit Guanosindiphosphat (GDP) und freiem Phosphat wird von der Succinyl-CoA-Synthetase katalysiert und liefert das zum ATP analoge energiereiche Guanosintriphosphat (GTP).
Biosynthese und Stoffwechsel
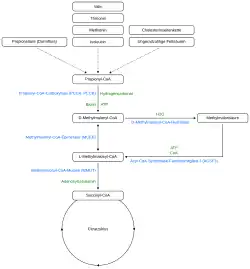
Methylmalonyl-CoA kann auf zwei Wegen synthetisiert werden:
- Aus Propionyl-CoA: Methylmalonyl-CoA entsteht im Körper bei der Umwandlung von Propionyl-CoA, das aus Fettsäuren mit ungerader Anzahl von C-Atomen entsteht, in Succinyl-CoA, das im Citratzyklus weiterreagiert. Die Bildungsreaktion aus Propionyl-CoA wird durch die Propionyl-CoA-Carboxylase in einer Biotin-abhängigen Carboxylierung katalysiert. Es entsteht zunächst das D-Enantiomer. Die Methylmalonyl-CoA-Racemase katalysiert die Isomerisierung von D-Methylmalonyl-CoA zu L-Methylmalonyl-CoA. Dieses Zwischenprodukt wird durch die L-Methylmalonyl-CoA-Mutase zum Succinyl-CoA umgesetzt. Die Reaktion benötigt Vitamin-B12 als Kofaktor.[2] Beim Abbau einiger Aminosäuren wie Isoleucin, Valin, Methionin und Threonin entsteht ebenfalls Propionyl-CoA, das über den identischen Abbauweg wie bei Fettsäuren über Methylmalonyl-CoA zu Succinyl-CoA umgesetzt wird.[3] Des Weiteren kann Propionyl-CoA auch aus Propionsäure gebildet werden, welche Bakterien im Darm produzieren.[4]
- Aus Methylmalonsäure: Das mitochondriale Enzym Acyl-CoA-Synthetase-Familienmitglied 3 (ACSF3) katalysiert die Thioesterifizierung von Methylmalonsäure mit Coenzym A (CoA), um Methylmalonyl-CoA zu bilden.[5]
Pathobiochemie
Methylmalonazidurie
Defekte der L-Methylmalonyl-CoA-Mutase oder ausgeprägter Vitamin-B12-Mangel können zur Methylmalonazidurie führen, die über eine Akkumulation von Methylmalonyl-CoA zu toxischen Effekten führt.[3] Methylmalonyl-CoA trägt auch als Donor für die pathologische posttranslationale Modifikation der Lysinmethylmalonylierung, vermutlich in stärkerem Maße als die Methylmalonsäure selbst, zur Erkrankung bei.[6] Unbehandelt kann die Erkrankung innerhalb kürzester Zeit zu schweren Schädigungen des Gehirns oder zum Tode führen.
Kombinierte Malon- und Methylmalonazidurie (CMAMMA)
Bei der kombinierten Malon- und Methylmalonazidurie (CMAMMA) führen Mutationen im ACSF3-Gen zu einer Funktionsstörung des mitochondrialen Enzyms Acyl-CoA-Synthetase-Familienmitglied 3 (ACSF3), wodurch die Umwandlung von Methylmalonsäure in Methylmalonyl-CoA und dessen Einspeisung in den Citratzyklus beeinträchtigt wird.[7][8] Die Folge ist eine Akkumulation von Methylmalonsäure, verringerte Spiegel von Methylmalonyl-CoA und eine im Vergleich zu gesunden Kontrollgruppen verminderte Lysinmethylmalonylierung.[6]
Einzelnachweise
- ↑ Dieser Stoff wurde in Bezug auf seine Gefährlichkeit entweder noch nicht eingestuft oder eine verlässliche und zitierfähige Quelle hierzu wurde noch nicht gefunden.
- ↑ a b H. Robert Horton, Laurence A. Moran, K. Gray Scrimgeour, J. David Rawn, Marc D. Perry: Biochemie. 4. Auflage, Pearson Education, 2008, ISBN 978-3-8273-7312-0, S. 674–675.
- ↑ a b c R. Witkowski, O. Prokop, E. Ullrich: Lexikon der Syndrome und Fehlbildungen: Ursachen, Genetik, Risiken. 7. Auflage, Springer, 2003, ISBN 978-3-540-44305-6, S. 818.
- ↑ Matthias R Baumgartner, Friederike Hörster, Carlo Dionisi-Vici, Goknur Haliloglu, Daniela Karall, Kimberly A Chapman, Martina Huemer, Michel Hochuli, Murielle Assoun, Diana Ballhausen, Alberto Burlina, Brian Fowler, Sarah C Grünert, Stephanie Grünewald, Tomas Honzik, Begoña Merinero, Celia Pérez-Cerdá, Sabine Scholl-Bürgi, Flemming Skovby, Frits Wijburg, Anita MacDonald, Diego Martinelli, Jörn Oliver Sass, Vassili Valayannopoulos, Anupam Chakrapani: Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. In: Orphanet Journal of Rare Diseases. Band 9, Nr. 1, Dezember 2014, doi:10.1186/s13023-014-0130-8, PMID 25205257, PMC 4180313 (freier Volltext).
- ↑ NIH Intramural Sequencing Center Group, Jennifer L Sloan, Jennifer J Johnston, Irini Manoli, Randy J Chandler, Caitlin Krause, Nuria Carrillo-Carrasco, Suma D Chandrasekaran, Justin R Sysol, Kevin O'Brien, Natalie S Hauser, Julie C Sapp, Heidi M Dorward, Marjan Huizing, Bruce A Barshop, Susan A Berry, Philip M James, Neena L Champaigne, Pascale de Lonlay, Vassilli Valayannopoulos, Michael D Geschwind, Dimitar K Gavrilov, William L Nyhan, Leslie G Biesecker, Charles P Venditti: Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. In: Nature Genetics. Band 43, Nr. 9, September 2011, ISSN 1061-4036, S. 883–886, doi:10.1038/ng.908, PMID 21841779, PMC 3163731 (freier Volltext) – (nature.com).
- ↑ a b PamelaSara E. Head, Sangho Myung, Yong Chen, Jessica L. Schneller, Cindy Wang, Nicholas Duncan, Pauline Hoffman, David Chang, Abigael Gebremariam, Marjan Gucek, Irini Manoli, Charles P. Venditti: Aberrant methylmalonylation underlies methylmalonic acidemia and is attenuated by an engineered sirtuin. In: Science Translational Medicine. Band 14, Nr. 646, 25. Mai 2022, ISSN 1946-6234, doi:10.1126/scitranslmed.abn4772, PMID 35613279, PMC 10468269 (freier Volltext) – (science.org).
- ↑ Caitlyn E. Bowman, Michael J. Wolfgang: Role of the malonyl-CoA synthetase ACSF3 in mitochondrial metabolism. In: Advances in Biological Regulation. Band 71, Januar 2019, S. 34–40, doi:10.1016/j.jbior.2018.09.002, PMID 30201289, PMC 6347522 (freier Volltext).
- ↑ Marie Cosette Gabriel, Stephanie M. Rice, Jennifer L. Sloan, Matthew H. Mossayebi, Charles P. Venditti, Huda B. Al‐Kouatly: Considerations of expanded carrier screening: Lessons learned from combined malonic and methylmalonic aciduria. In: Molecular Genetics & Genomic Medicine. Band 9, Nr. 4, April 2021, doi:10.1002/mgg3.1621, PMID 33625768, PMC 8123733 (freier Volltext).
Literatur
- G. Löffler, P. E. Petrides, P. C: Heinrich: Biochemie und Pathobiochemie. 8. Auflage, Springer, Heidelberg 2006, ISBN 978-3-540-32680-9.